Research/ VDD
Symmetric Computing has developed the Virtual Drug Discovery Platform (VDDP) which is a software pipeline that uses a combination of ligand docking and molecular dynamics to identify small molecule drug candidates. The VDDP is hosted on our Ada hybrid CPU/GPU supercomputer. It includes a database of nearly two billion small molecules with their 3d structures, a database of proteins with their 3d Xray crystal structures from human and other organisms, and a database of over ten thousand experimental and deployed drugs along with their target proteins and binding sites. Using this platform we are able to conduct early stage drug discovery research that will significantly reduce the time and cost of identifying lead compounds.
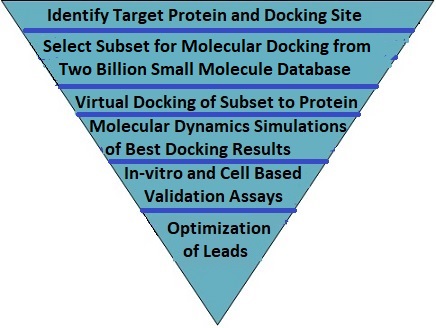
The Virtual Drug Discovery Platform supports high throughput vitual screening of millions of small molecules for binding to a target protein. We screen compounds from our database or from proprietary databases and select the best candidates for lead compounds. This is illustrated in the diagram and workflow procedure below.
Procedure
| 1. | Select the screening subset from the full small molecule database. This is done by examining the physical characteristics of the target protein binding site. The small molecule database is then filtered by molecular weight, solubility, pH and charge. Additional filtering may be done based on homologies determined by AI programs. |
| 2. | Execute docking of each small molecule in the screening subset to the target protein. Select a subset of candidates based on a specified binding affinity cutoff and maximum subset selection size. |
| 3. | Perform a molecular dynamics simulation of the protein - small molecule ligand complex for each of the molecules in the selected subset. Based on the free energy and disassociation constant scores, select the best lead compounds. |
A key requirement of virtual screening is that the binding sites are known for the target protein. The standard process for determining binding sites involves site directed mutagenesis requiring three to six months of laboratory work. Symmetric Computing provides an alternate method utilizing computer modeling techniques. We elucidate potential binding sites by calculating the solvent accessible volumes of the protein surface cavities. We then utilize a combination of geometric docking and molecular dynamics to characterize a binding site. This procedure is repeated for all target surface cavities that meet the proper criteria for protein binding pockets. In this manner we can identify both active and allosteric binding sites within days.
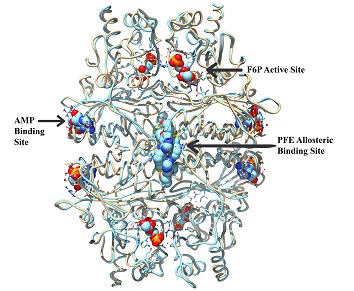
A major source of failure of lead compounds is the occurence of unanticipated side effects (drug toxicity) due to interactions with off-target proteins. Symmetric Computing addresses this problem by allowing virtual screening of selected lead compounds against our database of human proteins with 3d structures. In this manner we can identify many highly toxic lead compounds before proceeding with vivo validation.
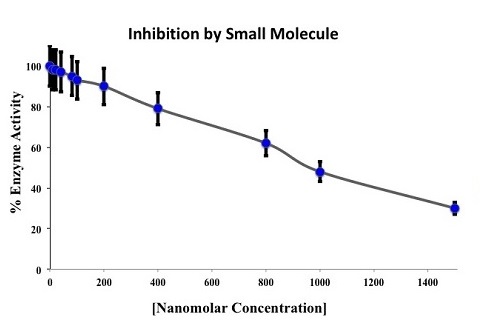
Symmetric Computing validates lead compounds selected through the VDDP virtual screening process using colorimetric, fluorometric or NMR spectral techniques. The IC50, and Ki or Kd for the target protein-ligand complex is then determined using these methods.
Symmetric Computing can provide capabilities that support drug discovery, such as DNA sequence assembly, construction of custom databases and 3D modeling of small molecules, DNA oligomers, peptides and other compounds, including their electrostatic maps.